 Self-assembly of amphiphile molecules is one
of current key subjects
in various research fields such as nano-scale technology, biotechnology
and molecular science. The ordering process of self-assembled monolayers
(SAMs) of alkanethiolate, particularly on the (111) surface of gold, has
been extensively studied as the simplest prototypical SAM system to understand
the self-assembly mechanism [1,2]. It is widely known that the Au-thiolate
interface structure on the Au(111) surface is similar to the hexagonal molecular
lattice in the (001) plane of single-crystal bulk alkane, leading to facile formation
of strain-free well-ordered monolayers. In case of Cu(100), however, there is obvious
difference in structure between the four-fold-symmetry surface and the molecular packing
in the bulk alkane, which will cause significant lattice mismatch between the Cu-thiolate
interface and the alkyl-chain layer. The goal of this study is to elucidate how the
alkanethiolate molecules reconcile the lattice mismatch to form a self-assembled monolayer.
This will be quite important to understand the self-assembly mechanism because this kind of
situation is rather general for actual molecular self-assembling systems.
STM observations for a hexanethiolate monolayer adsorbed on Cu(100) revealed that two-dimensionally (2D) ordering process needs a much longer period (more than 10 hours) compared to that on Au(111). This enables us to trace the self-assembling process by using surface XAFS technique. S-K and C-K XAFS spectra were measured at BL-11B and BL-7A, respectively, for the hexanethiolate monolayer [3]. Figure 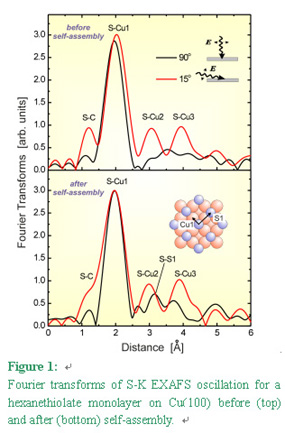 1 shows Fourier transforms of S-K EXAFS oscillations for the hexanethiolate monolayer measured
before (top) and after self-assembly (bottom). Quantitative analyses for S-Cu shells indicated that
the sulfur atom of the thiolate is located at the four-fold hollow site of the unreconstructed
Cu(100) surface irrespective of self-assembly. Although no drastic change was observed after self-assembly,
it is appreciable that a new peak appears at around 3.2 Å in the normal incidence curve, which
is associated with contribution from the nearest-neighbor (NN) sulfur atoms. Curve-fitting analysis
for this peak reveals that the NN sulfur atoms are located at a distance of c(2x2) lattice (S1 in the
Inset). The absence of the 3.2 Åpeak before self-assembly implies poor ordering at the Cu-S interface,
though C-K NEXAFS shows standing-up alignment of the alkyl chains even just after adsorption. Thus,
surface thiolates rapidly form a well-aligned but 2D-disordered monolayer with randomly occupying the
hollow sites. This rapid process is followed by slow evolution of the 2D-ordered structure via.
surface diffusion. Combination of the EXAFS results and force field calculations revealed that
a large lattice mismatch between the S/Cu layer and the alkyl-chain layer (31 %max) is effectively
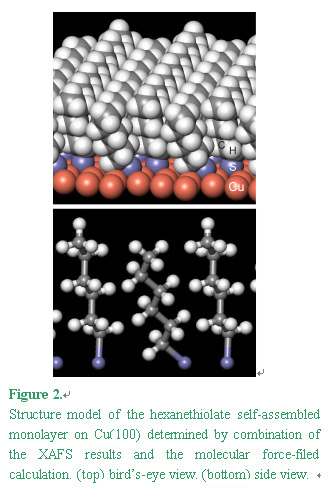
reduced (7 % max) by tilting half of the S-C bonds that bridges the two layers as shown in Figure 2.
The S-C bond tilting was detected as a decrease in polarization dependence of the S-C contribution
in the S-K XAFS spectra after self-assembly. The present XAFS study provides clear evidence for
the self-assembly mechanism, in which the lattice mismatch is effectively accommodated by the
internal degree of freedom of the molecule at the interface. 1 shows Fourier transforms of S-K EXAFS oscillations for the hexanethiolate monolayer measured
before (top) and after self-assembly (bottom). Quantitative analyses for S-Cu shells indicated that
the sulfur atom of the thiolate is located at the four-fold hollow site of the unreconstructed
Cu(100) surface irrespective of self-assembly. Although no drastic change was observed after self-assembly,
it is appreciable that a new peak appears at around 3.2 Å in the normal incidence curve, which
is associated with contribution from the nearest-neighbor (NN) sulfur atoms. Curve-fitting analysis
for this peak reveals that the NN sulfur atoms are located at a distance of c(2x2) lattice (S1 in the
Inset). The absence of the 3.2 Åpeak before self-assembly implies poor ordering at the Cu-S interface,
though C-K NEXAFS shows standing-up alignment of the alkyl chains even just after adsorption. Thus,
surface thiolates rapidly form a well-aligned but 2D-disordered monolayer with randomly occupying the
hollow sites. This rapid process is followed by slow evolution of the 2D-ordered structure via.
surface diffusion. Combination of the EXAFS results and force field calculations revealed that
a large lattice mismatch between the S/Cu layer and the alkyl-chain layer (31 %max) is effectively
reduced (7 % max) by tilting half of the S-C bonds that bridges the two layers as shown in Figure 2.
The S-C bond tilting was detected as a decrease in polarization dependence of the S-C contribution
in the S-K XAFS spectra after self-assembly. The present XAFS study provides clear evidence for
the self-assembly mechanism, in which the lattice mismatch is effectively accommodated by the
internal degree of freedom of the molecule at the interface.
References [1] P. Fenter et al. J. Chem. Phys. 106 (1997) 1600. [2] H. Kondoh et al. J. Chem. Phys. 111 (1999) 1175. [3] H. Kondoh, N. Saito, F. Matsui, T. Yokoyama, T. Ohta, and H. Kuroda, J. Phys. Chem. B 105 (2001) 12870. |